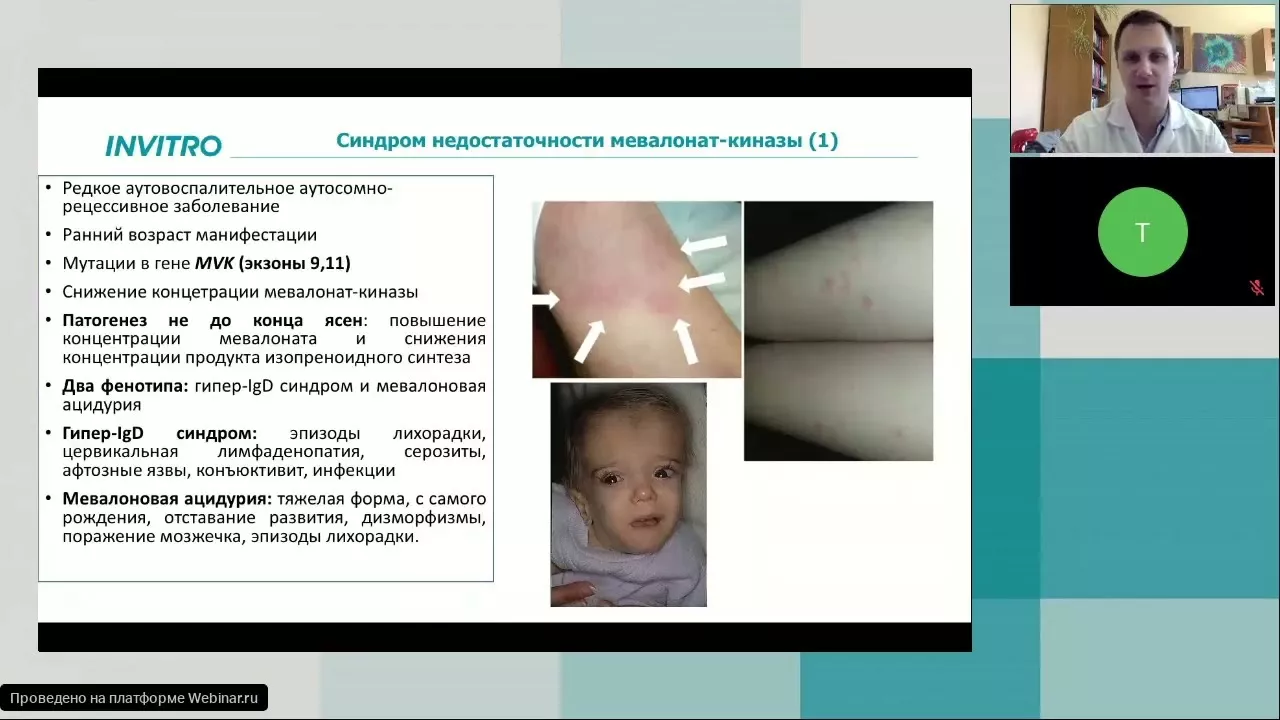
Определение
Периодический синдром, ассоциированный с рецептором ФНО (Tumor necrosis factor (TNF) Receptor-Associated Periodic Syndrome – TRAPS) или семейная ирландская лихорадка – это аутосомно-доминантно наследуемое заболевание, обусловленное мутацией гена рецептора I типа фактора некроза опухоли (ФНО) TNFRSF1A и характеризующееся приступами лихорадки, абдоминальной боли, интенсивной миалгии и болезненной эритемы на туловище или конечностях, обычно длящимися дольше 1 недели.
Эпидемиология
TRAPS впервые был описан в 1982 г. в большой ирландско-шотландской семье. Заболевание является орфанным и распространенность по некоторым данным составляет 1 : 1 000 000. Однако в популяции североевропейского происхождения частота может достигать 1:100 000. По данным международного регистра Eurofever/Eurotraps число больных TRAPS в 2013 г. составило 158 (105 взрослых и 53 ребенка). Соотношение мужчин и женщин среди больных TRAPS составляет 3:2.
Патогенез
Патогенез TRAPS сложен и включает множество механизмов. Связывание TNF c TNFR1 приводит к активации сигнального пути, который регулирует экспрессию генов провоспалительных цитокинов. В норме под воздействием ФНО-α внеклеточно расположенная часть TNFR1 подвергается расщеплению с последующим удалением с мембраны клетки. В результате увеличивается количество растворимых рецепторов ФНО, которые связывают данный цитокин во внеклеточной среде, предотвращая тем самым связывание ФНО рецепторами, расположенными на мембранах клеток, а значит и последующие провоспалительные эффекты. Мутировавшие рецепторы теряют способность уходить с поверхности клетки и стимулируют сигнальный путь NF-κB, что приводит к гиперпродукции ИЛ-1β, ИЛ-6, TNFα и хемокинов. Дефект удаления рецепторов ФНО лишь частично объясняет патогенез TRAPS-синдрома, так как при некоторых мутациях данный процесс не нарушен.
![Рис.1. Патогенетические механизмы TRAPS [2]](/upload/dev2fun.imagecompress/webp/medialibrary/02d/TRAPS.webp "Рис.1. Патогенетические механизмы TRAPS [2]")
Рис.1. Патогенетические механизмы TRAPS [2]
Тяжесть течения заболевания, так же, как и риск развития амилоидоза, определяется характером мутаций. Всего на сегодняшний день описано 58 мутаций в гене TNFRSF1A. Наиболее неблагоприятными являются миссенс-мутации, приводящие к замене цистеина во внеклеточной части рецептора TNFR1, следствием чего является исчезновение дисульфидных связей, как при мутации C88Y. Более распространенные варианты мутаций, такие как R92Q и P46L, обычно связаны с более мягкими клиническими фенотипами, а также могут быть выявлены у клинически здоровых лиц.
При мутациях, затрагивающих нецистеиновые остатки, снижается пенетрантность клинического фенотипа (82% против 93% при заменах цистеина), а риск амилоидоза уменьшается до 2% (против 24% при цистеиновых мутациях).
Учитывая, что применение при данном заболевании этанерцепта – антагониста ФНО, сопровождается быстрым наступлением стойкой ремиссии, предполагается, что и сам ФНО играет определенную роль в развитии этого синдрома.
Клиническая картина
Большинство пациентов заболевают в детском возрасте (средний возраст – 4 года), однако имеются случаи дебюта у взрослых. Возраст появления первых симптомов может значительно варьировать даже внутри одной семьи.
Частота и длительность воспалительных атак при TRAPS-синдроме могут быть различными. В среднем, они происходят каждые 6 недель и длятся дольше 1 недели.
Наиболее частые клинические проявления TRAPS включают: лихорадку (96%), боль в животе (70%), миалгию (69%), артралгию (69%), эритематозную сыпь (60%), острый конъюнктивит (37%), периорбитальный отек (28%).
Мигрирующая эритема обычно локализуется над областями миалгий и сохраняющаяся в течение 4–21 дня.
Атаки TRAPS могут провоцироваться эмоциональными и физическими стрессами, инфекциями, травмой, гормональной перестройкой, слабостью, вакцинацией. Однако, в большинстве случаев триггерный фактор остается неизвестным.
Со временем может развиться вторичный амилоидоз с почечными и печеночными проявлениями. Частота развития амилоидоза без лечения составляет 15-25%. Наибольший риск развития амилоидоза наблюдается у пациентов с патогенными вариантами, влияющими на цистеиновые остатки или носителей p.T79M (T50M) варианта, в результате более длительных приступов и более выраженного воспаления. Высокий уровень АА протеина в крови – индикатор отложения амилоида.
Диагностические критерии
Клинические классификационные критерии TRAPS Eurofever/PRINTO 2015 [3]|
Наличие: |
Баллы |
|---|---|
|
Периорбитальный отек |
21 |
|
Продолжительность эпизодов >6 дней |
19 |
|
Мигрирующая сыпь* |
18 |
|
Миалгия |
6 |
|
Родственники с этой патологией |
7 |
|
Отсутствие: |
Баллы |
|
Рвота |
14 |
|
Афтозный стоматит |
15 |
|
Пограничное значение (cut-off) |
≥ 43 |
Оцениваемые клинические признаки должны быть связаны с типичной атакой TRAPS (применение данных критериев возможно только после исключения интеркуррентных инфекций и коморбидных заболеваний). Критерии оцениваются путем суммирования баллов при наличии или отсутствии клинического признака, пороговым значением для постановки диагноза TRAPS является сумма ≥43 баллам.
Классификационные критерии TRAPS Eurofever/PRINTO 2019 г. при наличии молекулярно-генетического исследования [4]Диагноз TRAPS устанавливается при наличии подтвержденного генотипа* TNFRSF1A и как минимум одного из критериев, перечисленных ниже ИЛИ при отсутствии подтверждающего генотипа** TNFRSF1A и наличии как минимум двух критериев, перечисленных ниже:
-
Продолжительность приступов ≥ 7 дней
-
Миалгия
-
Мигрирующая сыпь
-
Периорбитальный отек
-
Наличие заболевания у родственников
*Патогенные или вероятно патогенные варианты (гетерозиготные при аутосомно- доминантном типе наследования болезни)
**Варианты неопределенной клинической значимости (VUS). Доброкачественные или вероятно доброкачественные варианты исключаются.
Пациент с подтвержденным повышением острофазовых показателей воспаления (СОЭ, или СРБ, или SAA) четко коррелирующих с клиническими проявлениями атаки при тщательном исключении возможных маскирующих болезней (онкологические, инфекционные, аутоиммунные и другие врожденные аномалии иммунного ответа), при длительности анамнеза заболевания не менее 6 мес. может расцениваться как пациент с наследственной периодической лихорадкой (в данном случае – TRAPS).
Лабораторная диагностика
Как и при других аутовоспалительных заболеваниях, в период приступов повышаются уровни острофазовых показателей (СОЭ, СРБ, SAA, фибриноген, гаптоглобин, ферритин). СОЭ может оставаться повышенной и в межприступном периоде. В общем анализе крови можно выявить анемию хронического заболевания, лейкоцитоз, тромбоцитоз. Кроме того, при TRAPS-синдроме может быть повышен уровень IgD, но не выше 100 ед/мл. Типичным является снижение уровня растворимого рецептора ФНО (TNFRSF1A) в сыворотке крови, как в период приступа, так и между атаками. Нормальный уровень растворимого TNFRSF1A не исключает диагноз. Окончательно диагноз подтверждается генетическими исследованиями, обнаруживающими мутации в гене TNFRSF1A.
Наша лаборатория выполняет комплексный тест направленный на выявление клинически сходных синдромов: дефицита мевалонат-киназы (MVK) и TRAPS-синдрома:
Также доступен расширенный тест, позволяющий проводить дифференциальную диагностику наиболее часто встречающихся аутовоспалительных заболеваний:
Чем TRAPS-синдром отличается от семейной средиземноморской лихорадки?
⌄Оба заболевания относятся к группе наследственных моногенных аутовоспалительных синдромов, но имеют принципиально разную природу. TRAPS-синдром отличается от семейной средиземноморской лихорадки (FMF) длительностью приступов, генетическим механизмом наследования, специфическими кожными проявлениями и ответом на стандартную терапию.
Длительность приступа:
-
TRAPS: длительные атаки, длящиеся от 1 до 3 недель.
-
FMF: короткие атаки, длящиеся от 1 до 3 дней.
Генетическая причина:
-
TRAPS: патогенны варианты в гене TNFRSF1A.
-
FMF: патогенны варианты MEFV.
Тип наследования:
-
TRAPS: аутосомно-доминантный (передается от больного родителя).
-
FMF: аутосомно-рецессивный (родители обычно здоровые носители).
Этническая привязка:
-
TRAPS: встречается у любых национальностей во всем мире.
-
FMF: чаще болеют народы Средиземноморья (армяне, турки, евреи-сефарды, арабы).
Характер сыпи:
-
TRAPS: болезненные мигрирующие пятна, перемещающиеся по телу.
-
FMF: рожеподобная сыпь, четко локализованная на голенях и стопах.
Мышечные боли:
-
TRAPS: интенсивные миалгии, которые «перемещаются» вместе с сыпью.
-
FMF: миалгии не характерны.
Особые симптомы:
-
TRAPS: типичны отеки вокруг глаз (периорбитальные) и конъюнктивит.
-
FMF: типичен острый перитонит (сильнейшие боли в животе) и плеврит.
Как подтвердить диагноз TRAPS-синдрома?
⌄Для подтверждения TRAPS-синдрома используется молекулярно-генетическое исследование (выявление патогенных вариантов в гене TNFRSF1A). Диагноз критериальный и учитывает как клинические данные, так и данные молекулярно-генетического исследования.
1) Периодический синдром, ассоциированный с мутацией гена рецептора фактора некроза опухоли (TRAPS) (Другие уточненные нарушения с вовлечением иммунного механизма, не классифицированные в других рубриках). Клинические рекомендации МЗ РФ, 2023 г.
2) Gattorno, M. (2019). The TNF Receptor-Associated Autoinflammatory Syndrome (TRAPS). In: Efthimiou, P. (eds) Auto-Inflammatory Syndromes. Springer, Cham. https://doi.org/10.1007/978-3-319-96929-9_3
3) Federici S, Sormani MP, Ozen S et al. Paediatric Rheumatology International Trials Organisation (PRINTO) and Eurofever Project. Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis. 2015 May;74(5):799-805. doi: 10.1136/annrheumdis-2014-206580
4) Gattorno M, Hofer M, Federici S et al. Eurofever Registry and the Paediatric Rheumatology International Trials Organisation (PRINTO). Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. 2019 Aug;78(8):1025-1032. doi: 10.1136/annrheumdis-2019-215048