Определение
Аутовоспалительные заболевания (АВЗ) или аутовоспалительные синдромы (АВС) – это группа заболеваний, характеризующихся немотивированными приступами системного воспаления, проявляющегося полиморфной клинической симптоматикой, которая может включать лихорадку, сыпь, артриты, серозиты, поражение ЦНС и другие симптомы при отсутствии аутоиммунных и инфекционных причин.
Патофизиология аутовоспаления


- CAPS - криопирин-ассоциированные периодические синдромы (мутация NLRP3)
- DIRA - дефицит антагониста рецептора IL -1 (мутация IL-1RA)
- DITRA - дефицит антагониста рецептора IL -36 (мутация IL-36RA)
- FMF - наследственная средиземноморская лихорадка (мутация в гене пирина)
- HA20 - гаплонедостаточность A20 (мутация TNFAIP3, который в норме ингибирует эндотоксин- и TNF-индуцированную активацию NF-кB)
- PAPA - PAPA-синдром (гнойный артрит, гангренозная пиодермия и акне), вызван мутацией в гене CD2BP1 (PSTPIP1)
- PRAAS - протеасомно-ассоциированные аутовоспалительные синдромы (PSMB8 – один из генов, мутации в котором приводят к развитию PRAAS)
- SAVI - STING-ассоциированная васкулопатия с ранним началом (мутация в STING)
- TRAPS - периодический синдром, ассоциированный с рецептором ФНО (мутация в TNFR1)
Различия между аутоиммунными и аутовоспалительными заболеваниями
|
|
аутоиммунные заболевания |
аутовоспалительные заболевания |
|---|---|---|
|
нарушения в иммунной системе затрагивают… |
адаптивный иммунитет; чаще всего органоспецифичны |
врождённый иммунитет; не органоспецифичны |
|
генетика |
часто полигенные |
часто моногенные |
|
преобладающие эффекторные механизмы и клетки |
В-клетки → ауто-АТ; аутореактивные Т-клетки |
ИЛ-1β и др.провоспалит. цитокины; моноциты, макрофаги, нейтрофилы |
|
патогенез поражения органов |
ауто-АТ- или ауто-АГ-специфический, опосредованный Т-клетками |
опосредован нейтрофилами и макрофагами |
|
главные терапевтические цели и препараты, воздействующие на них |
B- или Т-клетки (ритуксимаб, алемтузумаб) |
ИЛ-1β, ИЛ-6, ФНО-α (анакинра, тоцилизумаб, этанерцепт) |
Классификация аутовоспалительных синдромов
Мы приводим классификацию EUROFEVER [3] как наиболее авторитетную.
- Семейная средиземноморская лихорадка (аутосомно-рецессивное наследование)
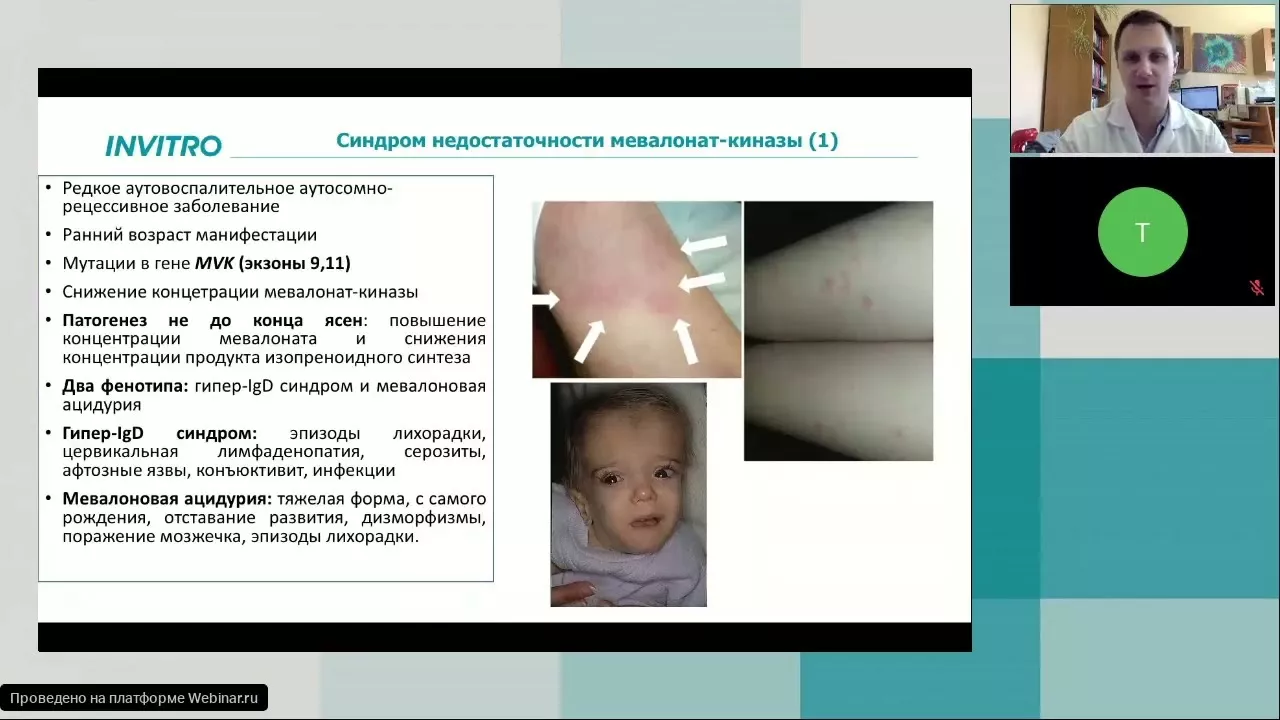
- Синдром дефицита мевалонаткиназы (аутосомно-рецессивное наследование)
- Периодический синдром, ассоциированный с рецептором ФНО (TRAPS) (аутосомно-доминантное наследование)
Это моногенные заболевания, которые характеризуются рецидивирующими вспышками системного воспаления, проявляющимися внезапными приступами лихорадки, сопровождающимися резким повышением острофазовых тестов и рядом клинических проявлений, таких как сыпь, серозит (перитонит, плеврит), лимфаденопатия, артриты. Вспышки заболевания обычно разделены бессимптомными интервалами различной продолжительности, характеризующимися полным благополучием, нормальным ростом и полной нормализацией острофазовых тестов.
- PFAPA-синдром или синдром Маршалла (периодическая лихорадка, афтозный стоматит, фарингит и шейный лимфаденит), является спорадическим, не наследственным синдромом.
2) NLRP-ассоциированные аутовоспалительные заболевания (наследственные криопирин-ассоциированные периодические синдромы или криопиринопатии):
- семейный холодовой аутовоспалительный синдром,
- синдром Макла-Уэльса
- младенческое мультисистемное воспалительное заболевание (хронический младенческий нервно-кожно-суставной синдром)
- Синдром Блау (характеризуется неказеозным гранулематозным воспалением с поражением суставов, кожи и сосудистой оболочки глаз и связан с мутациями гена CARD15 (NOD2)). Наследуется аутосомно-доминантно.
- PAPA-синдром (гнойный артрит, гангренозная пиодермия и акне), вызван мутацией CD2BP1 (PSTPIP1). Наследуется аутосомно-доминантно.
- Синдром Маджида (характеризуется хроническим рецидивирующим многоочаговым остеомиелитом, врожденной дизэритропоэтической анемией и нейтрофильным дерматозом; вызван мутациями гена LPIN2). Наследуется аутосомно-рецессивно.
- Дефицит антагониста рецептора интерлейкина-1 (DIRA-синдром) (характеризуется дебютом в неонатальном периоде, мультифокальным остеомиелитом, периоститом и кожными пустулами, вызван мутацией в гене IL1RN). Наследуется аутосомно-рецессивно.
- Дефицит антагониста рецептора IL-36 (DITRA-синдром). Характерны внезапные эпизоды гнойничковой сыпи, сопровождающиеся высокой температурой и недомоганием. Вызван мутациями в гене IL36RN. Наследуется аутосомно-рецессивно.
5) Протеасомно-ассоциированные аутовоспалительные синдромы (PRAAS)
- Хронический атипичный нейтрофильный дерматоз с липодистрофией и подъемами температуры (CANDLE-синдром),
- Синдром с контрактурами суставов, атрофией мышц, микроцитарной анемией и панникулит-индуцированной липодистрофией (JMP),
- Синдром Накаджо-Нишимура (NNS)
- Японский аутовоспалительный синдром с липодистрофией (JASL)
Эти аутосомно-рецессивные заболевания чаще всего вызваны мутациями в гене PSMB8, который кодирует субъединицу β5i протеосом. Также могут быть вызваны мутациями в генах PSMB4, PSMB9, PSMA3, POMP. PRAAS манифестируют в младенчестве. Отмечаются рецидивирующая лихорадка, сыпь в виде кольцевидных бляшек, дактилит, артрит и повышенные маркеры воспаления. Могут быть гепатомегалия, спленомегалия, мышечные атрофии, лимфаденопатия, отставание в развитии, контрактуры суставов и липодистрофия, а также ранняя смерть, связанная с полиорганной недостаточностью.
Патогенетическая классификация моногенных системных аутовоспалительных заболеваний [2]
|
Заболевание |
Ген |
Характер наследования |
|---|---|---|
|
1. IL-1β-опосредованные заболевания |
||
|
1.1. Семейные лихорадки (моногенные) |
||
|
Семейная средиземноморская лихорадка |
MEFV |
Аутосомно-рецессивный |
|
Криопирин-ассоциированные периодические синдромы (CAPS): FCAS/MWS/CINCA (NOMID) |
NLRP3 |
Аутосомно-доминантный |
|
Гипер-IgD синдром (HIDS) |
MVK |
Аутосомно-рецессивный |
|
PFAPA-синдром или синдром Маршалла |
- |
Спорадический (не наследственный) |
|
1.2. Другие моногенные синдромы |
||
|
DIRA-синдром (дефицит антагониста рецептора ИЛ-1) |
IL1RN |
Аутосомно-рецессивный |
|
Синдром Маджида |
LPIN2 |
Аутосомно-рецессивный |
|
PAPA-синдром (гнойный артрит, гангренозная пиодермия и акне) |
PSTPIP1 |
Аутосомно-доминантный |
|
1.3. Пиогенные / приобретённые заболевания |
||
|
Синдром SAPHO (синовит, акне, пустулез, гиперостоз и остеит) |
- |
- |
|
Синдром Шницлера |
Полигенный / NLRP3 |
De novo |
|
PAPASH-синдром (гнойный артрит, акне, гангренозная пиодермия и гнойный гидраденит) |
PSTPIP1 |
Неизвестен |
|
PASH-синдром (гангренозная пиодермия, акне и гнойный гидраденит) |
PSTPIP1, MEFV, NOD2, IL1RN, PSMB8 |
Неизвестен |
|
2. IL-36-опосредованные заболевания |
||
|
DITRA-синдром (дефицит антагониста рецептора IL-36) |
IL36RN |
Аутосомно-рецессивный |
|
3. NF-kB-опосредованные заболевания |
||
|
Синдром Блау / саркоидоз с ранним началом |
NOD2 (CARD15) |
Аутосомно-доминантный, de novo |
|
CAMPS-синдром (CARD14-опосредованный псориаз) |
CARD14 |
Аутосомно-доминантный |
|
ORAS-синдром (OTULIN-ассоциированный аутовоспалительный синдром) |
OTULIN |
Аутосомно-рецессивный |
|
4. TNFα-опосредованные заболевания |
||
|
Бехчета-подобный семейный аутовоспалительный синдром (AISBL) или синдром гаплонедостаточности А20 (HA20) |
TNFAIP3 |
Аутосомно-доминантный |
|
TRAPS-синдром (периодический синдром, ассоциированный с рецептором ФНО) |
TNFRSF1A |
Аутосомно-доминантный |
|
5. Интерферон-опосредованные заболевания |
||
|
DADA-2-синдром (дефицит аденозиндезаминазы 2) |
ADA-2 (CECR1) |
Аутосомно-рецессивный |
|
PLCG2-ассоциированные синдромы |
PLCG2 |
Аутосомно-доминантный |
|
PRAАS (протеасомно-ассоциированный аутовоспалительный синдром) |
PSMB8 |
Аутосомно-рецессивный |
|
SAVI-синдром (STING-ассоциированная васкулопатия с ранним началом) |
TMEM173 (STING1) |
Аутосомно-доминантный |
Лабораторная диагностика аутовоспалительных заболеваний
Золотым стандартом является генетическое обследование на наличие специфических мутаций. В нашей лаборатории имеется спектр генетических тестов для диагностики наиболее частых форм наследственных моногенных АВЗ:
Тест 01.02.15.1175. Семейная средиземноморская лихорадка. Выявление мутаций в гене MEFV.
Тест 01.02.15.1620. Исследование при криопирин-ассоциированных периодических синдромах (ген NLRP3)
Тест 01.02.05.255. Диагностика редких аутовоспалительных синдромов (дефицит мевалонат-киназы (MVK), TRAPS-синдром)
- Diprose WK, Jordan A, Anderson NE. Autoinflammatory syndromes in neurology: when our first line of defence misbehaves. Pract Neurol. 2022 Apr;22(2):145-153. doi: 10.1136/practneurol-2021-003031
- Meier-Schiesser B, French LE. Autoinflammatory syndromes. J Dtsch Dermatol Ges. 2021 Mar;19(3):400-426. DOI: 10.1111/ddg.14332
- Eurofever Project - What are Autoinflammatory diseases?
- Kul Cinar O, Putland A, Wynne K, Eleftheriou D, Brogan PA. Hereditary Systemic Autoinflammatory Diseases: Therapeutic Stratification. Front Pediatr. 2022 Apr 28;10:867679. doi: 10.3389/fped.2022.867679
- Efthimiou, P. (eds) Auto-Inflammatory Syndromes. Springer, Cham. https://doi.org/10.1007/978-3-319-96929-9
- Fatma Dedeoglu, Susan Kim, 14 - Autoinflammatory Disorders, Editor(s): Donald Y.M. Leung, Stanley J. Szefler, Francisco A. Bonilla, Cezmi A. Akdis, Hugh A. Sampson, Pediatric Allergy: Principles and Practice (Third Edition), Elsevier, 2016, Pages 133-142.e3, ISBN 9780323298759. https://doi.org/10.1016/B978-0-323-29875-9.00014-8.
- Rood JE, Behrens EM. Inherited Autoinflammatory Syndromes. Annu Rev Pathol. 2022 Jan 24;17:227-249. DOI: 10.1146/annurev-pathmechdis-030121-041528