Синонимы: наследственный семейный амилоидоз без невропатии, пароксизмальный синдром Джэйнуэя-Мозенталя, периодический перитонит, синдром Рейманна, болезнь Сигала-Маму
Определение
Семейная средиземноморская лихорадка (ССЛ) – наиболее часто встречающийся наследственное моногенное аутовоспалительное заболевание, распространённое преимущественно у жителей/выходцев из Средиземноморского региона и возникающее в результате миссенс-мутаций гена MEFV (кодирует белок пирин). Заболевание проявляется периодически возникающими немотивированными приступами лихорадки и сопровождается сильными болями в животе и/или грудной клетке и другой симптоматикой, в сочетании со значительным повышением уровня острофазовых маркеров в течение 12 - 72 часов. Важнейшим поздним осложнением заболевания является вторичный амилоидоз.
Эпидемиология
ССЛ распространена преимущественно у представителей народностей, предки которых жили на территории Средиземноморского бассейна (вне зависимости от места их нынешнего проживания), особенно у армян, евреев неашкеназского происхождения (чаще сефардов), турок, арабов и лишь в 6% случаев у лиц прочих национальностей. Распространенность заболевания в данной популяции может варьировать от 1:500 до 1:1000. По некоторым данным, каждый четвертый представитель средиземноморских этнических групп может являться носителем одной из мутаций гена MEFV.
Реже заболевание может встречаться у представителей других этносов: народов Закавказья (азербайджанцы, грузины), Северного Кавказа, Южной Европы (итальянцы, греки, испанцы), Балканского полуострова. Описаны случаи ССЛ у японцев, коренного населения Индии, цыган и других народов. Есть описания болезни у русских.
У взрослых ССЛ чаще встречается у мужчин, чем у женщин, соотношение 1,5–2:1. На течение болезни у женщин влияют половые гормоны – нередко приступы болезни возникают во время менструаций, отсутствуют во время беременности и возобновляются после родов. Риск развития амилоидоза почек выше у мужчин.
Этиология и патогенез
В основе патогенеза заболевания лежит миссенс-мутация в гене MEditerranean FeVer (MEFV), состоящем из 10 экзонов, расположенных на коротком плече 16 хромосомы (16p13.3). Ген MEFV кодирует белок пирин/маренострин, который играет важную роль в регуляции воспалительных механизмов. Данный белок в основном экспрессируется в моноцитах, нейтрофилах и в меньшей степени в дендритных клетках, коже и синовиальных фибробластах.
Традиционно характер наследования при ССЛ характеризуется как аутосомно-рецессивный [1], при этом для развития заболевания необходимо наличие носительства двух патогенных мутаций на парных хромосомах: одинаковых – гомозиготность или разных мутаций – компаунд-гетерозиготность. Однако, заболевание может развиться и у носителей одной патогенной мутации – гетерозигот. В этом случае характер наследования можно трактовать как аутосомно-ко-доминантное с неполной пенентрантностью. Доля гетерозигот среди больных ССЛ в различных популяциях составляет от 16,5% до 33,8%. Примерно у 10% пациентов с достоверным диагнозом ССЛ мутации гена MEFV вообще не обнаруживаются [1].
К настоящему времени известно 244 варианта мутаций гена MEFV.
Большинство известных патогенных мутаций локализуются в 10-м экзоне гена. Ряд мутаций локализован в экзонах 2, 3, 5. Наиболее распространенными мутациями являются p.M680I, p.M694V, p.M694I, pK695R, pV726A, pA744S и pR761H [1].
Мутации различаются по экспрессивности (патогенности). Наиболее экспрессивной мутацией, обуславливающей тяжесть течения и высокий риск неблагоприятного исхода, является мутация p.M694V. Она ассоциирована с тяжелым течением заболевания и высоким риском развития амилоидоза.
Данные о корреляциях генотипа и фенотипа и прочая информация о заболевании аккумулируется в международном регистре Eurofever
Большинство проявлений, которые в настоящее время приписываются ССЛ, связаны с изменением функции моноцитов и нейтрофилов. Основным механизмом развития заболевания является снижение ингибирующей активности мутантного пирина, приводящее к неконтролируемой гиперпродукции ИЛ-1β, что приводит к эпизодам воспаления (с сопутствующей лихорадкой) в брюшине, плевре и суставах.
В норме пирин связывает белок ASC, что, в свою очередь, предотвращает активацию каспазы-1. Каспаза-1 повышает синтез ИЛ-1β, активирует нуклеарный фактор каппа В и ИЛ-4-индуцированный апоптоз клеток.

У пациентов с ССЛ имеется аномально повышенная чувствительность к бактериальному эндотоксину, и транзиторная бактериемия (в норме – практически безвредное состояние) приводит к значительному повышению синтеза ИЛ-1β с развитием системного воспалительного ответа. Другими индукторами ИЛ-1β при ССЛ могут быть чрезмерные физические нагрузки, эмоциональное напряжение, изменение гормонального фона (у женщин).
Атаки ССЛ также характеризуются высвобождением внеклеточных ловушек нейтрофилов (NET), представляющих собой трехмерную сеть, образованную хроматиновыми филаментами, с расположенными на ней гранулярными и цитоплазматическими белками, включая активный ИЛ-1β. NET ограничивает свою генерацию путем механизма отрицательной обратной связи, что объясняет периодичность лихорадок.
Под действием провоспалительных цитокинов в печени повышается синтез белков острой фазы воспаления, в т. ч. сывороточного амилоида А, продукты деградации которого откладываются во внутренних органах. Развитие амилоидоза почек – наиболее серьезное позднее осложнение ССЛ, определяющее прогноз заболевания. Однако стоит отметить, что амилоидоз развивается только у пациентов с определёнными гаплотипами MEFV .
Клиническая картина
Первые признаки заболевания чаще всего появляются в детском возрасте: у 90% пациентов – до 20 лет жизни, у 60% – до 10-летнего возраста. Чем раньше манифестирует заболевание, тем тяжелее оно протекает.
Проявления семейной средиземноморской лихорадки можно разделить на два фенотипа: ССЛ 1-го и 2-го типа. ССЛ 1-го типа характеризуется приступами лихорадки и серозита. При ССЛ 2-го типа амилоидоз возникает как первое и единственное проявление болезни.
- Эпизоды лихорадки 38°- 40 °C и выше продолжительностью 12–72 ч (наблюдается у 100% больных). Проходят спонтанно. Отсутствует реакция на антибиотики. Возможные триггеры приступа - стресс, переохлаждение, жирная пища, инфекции, некоторые лекарства и менструальный цикл. Частота атак различна (у некоторых пациентов очень часто - раз в неделю, у некоторых довольно редко – раз в несколько лет), чаще всего интервал между атаками – 3–4 недели;
- Асептический перитонит, проявляющийся как острая боль в животе (82–98%);
- Плеврит, проявляющийся как односторонняя боль в груди, усиливающаяся при дыхании, кашель, одышка, поверхностное частое дыхание (30-40%);
- Артрит: как правило, моноартрит коленного, реже – тазобедренного или голеностопного сустава со значительным выпотом (75%);
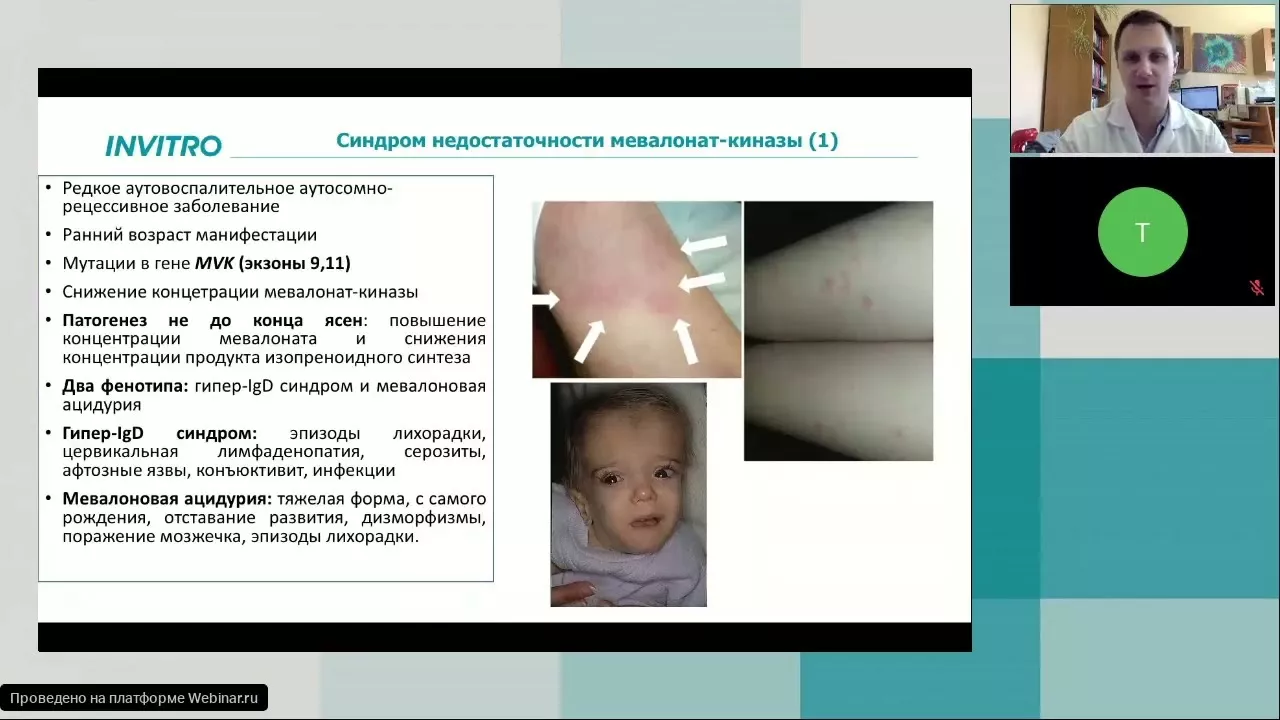
- Эризипелоидподобная (рожеподобная) сыпь (7-40%): отечные гиперемированные болезненные участки кожи 10–15 см в диаметре обычно локализуются ниже колена на передней или задней поверхности ноги – Рис 2.
- Неврологическая симптоматика: головная боль, светобоязнь, слезотечение, нарушение сна, шум в голове и ушах
- Миалгии (выраженные боли в мышцах)
- Отек и болезненность мошонки у молодых мужчин (5%): поражение, как правило, одностороннее, характеризуется болезненностью, отечностью и гиперемией мошонки на пораженной стороне, проходит самостоятельно по окончании приступа
- Перикардит (<1%)
![Эризипелоидподобная (рожеподобная) сыпь на спине (а) и на ногах (b) у пациентки с семейной средиземноморской лихорадкой [6].](/upload/dev2fun.imagecompress/webp/medialibrary/70c/FMF-skin-manifestation.webp "Эризипелоидподобная (рожеподобная) сыпь на спине (а) и на ногах (b) у пациентки с семейной средиземноморской лихорадкой [6].")
Рис 2. Эризипелоидподобная (рожеподобная) сыпь на спине (а) и на ногах (b) у пациентки с семейной средиземноморской лихорадкой [6].
Осложнения
Основным осложнением периодической болезни является АА-амилоидоз, как правило, локализующийся в почках. Иногда может отмечаться амилоидоз ЖКТ, печени, селезёнки, реже - сердца, яичек, щитовидной железы.
Амилоид А является продуктом распада сывороточного амилоида А (SAA). Это белок острой фазы воспаления, который вырабатывается в печени под действием ИЛ-1.
Риск развития амилоидоза составляет 25-70% (в зависимости от популяции). Риск максимален у пациентов, имеющих в числе проявлений артрит, гомозиготных по мутации p.M694V и имеющих α/α вариант гена SAA1. При своевременной постановке диагноза и своевременном назначением терапии риск амилоидоза составляет <1%.
Редкими осложнениями являются спаечная болезнь, развивающаяся в результате периодически повторяющегося перитонита, а также женское бесплодие, вызванное спаечной болезнью
Критерии диагностики семейной средиземноморской лихорадки
Для установления диагноза «семейная средиземноморская лихорадка» разработаны несколько вариантов диагностических критериев.
1. Диагностические критерии ССЛ Тель-Хашомер
|
Большие критерии |
Типичные атаки: 1. Перитонит (генерализованный) 2. Плеврит (односторонний) или перикардит 3. Моноартрит (коленного,тазобедренного, голеностопного суставов) |
|---|---|
|
Малые критерии |
1–3. Неполная атака с вовлечением ≥1 из нижеприведенных локализаций:
|
|
Дополнительные критерии |
1. Наличие случаев ССЛ в семейном анамнезе 2. Принадлежность к соответствующей этнической группе 3. Возраст начала заболевания до 20 лет 4–9. Характеристика атаки: 4. Тяжелая, приковывающая к постели 5. Спонтанное разрешение атаки 6. Наличие бессимптомных промежутков 7. Повышение уровня лабораторных маркеров воспаления с возрастанием значений ≥1 из следующих признаков: количество лейкоцитов в клиническом анализе крови, СОЭ, сывороточный амилоид А и/или фибриноген 8. Эпизоды протеинурии/гематурии 9. Непродуктивная лапаротомия или удаление «белого» (без флегмонозного воспаления) аппендикса 10. Родители пациента состоят в близкородственном браке |
Для постановки диагноза необходимо соответствие ≥1 большим критериям, или ≥2 малым критериям, или 1 большому + ≥5 дополнительным критериям, или 1 малому + ≥4 дополнительным критериям из числа первых 5 [1,2]
2. Турецкие педиатрические критерии ССЛ
-
Лихорадка (температура в подмышечной области 38,0оС, продолжительностью 6–72 часа, не менее 3-х эпизодов.
-
Боли в животе (продолжительность 6–72 часа, не менее 3-х эпизодов)
-
Боли в грудной клетке (продолжительность 6–72 часа, не менее 3-х эпизодов)
-
Артрит (продолжительность 6–72 часа, не менее 3-х эпизодов, олигоартрит)
-
Наличие пациентов с ССЛ в семье
Для постановки диагноза необходимо наличие 2 из 5 критериев [1]
3. Клинические диагностические критерии ССЛ (Eurofever 2015)
|
Наличие признака |
Баллы |
|---|---|
|
Продолжительность эпизодов <2 дней |
9 |
|
Боль в грудной клетке |
13 |
|
Абдоминальная боль |
9 |
|
Принадлежность к этносам Восточного Средиземноморья* |
22 |
|
Принадлежность к этносам Северного Средиземноморья** |
7 |
|
Отсутствие признака |
Баллы |
|
Афтозный стоматит |
9 |
|
Уртикарная сыпь |
15 |
|
Увеличение шейных лимфоузлов |
10 |
|
Продолжительность эпизодов >6 дней |
13 |
|
Пограничное значение (cut-off) |
≥ 60 |
|
*Этносы Восточного Средиземноморья: турки, армяне, неашкеназские евреи, арабы. **Этносы Северного Средиземноморья: итальянцы, испанцы, греки. |
|
Оцениваемые клинические признаки должны быть связаны с типичной атакой ССЛ (применение данных критериев возможно только после исключения интеркуррентных инфекций и коморбидных заболеваний). Критерии оцениваются путем суммирования баллов при наличии или отсутствии клинического признака, пороговым значением для постановки диагноза ССЛ является сумма ≥ 60 баллов [1-4]
4. Классификационные критерии Eurofever/PRINTO 2019 г. для ССЛ при наличии молекулярно-генетического исследования
Наличие подтверждающего генотипа* гена MEFV + минимум одного из следующих критериев ИЛИ
наличие неподтверждающего генотипа** гена MEFV + по меньшей мере 2-х из следующих критериев:
-
Длительность эпизодов – 1-3 дня
-
Артрит
-
Боли в грудной клетке
-
Боли в животе
Лабораторная диагностика
Во время приступов лихорадки в крови пациентов отмечается нейтрофильный лейкоцитоз со сдвигом формулы влево, повышение СОЭ и других острофазовых показателей (уровня С-реактивного белка, SAA-протеина, фибриногена, гаптоглобина, С3- и С4-фракций комплемента). Возможно развитие анемии хронического воспаления. В общем анализе мочи во время эпизодов лихорадки может отмечаться транзиторная гематурия и небольшая протеинурия. Если же уровень экскреции белка превышает 0,5 г/сут., то это может свидетельствовать в пользу амилоидоза почек.
Стандартом диагностики семейной средиземноморской лихорадки является молекулярно-генетическое исследование, подтверждающее наличие патогенных вариантов в гене MEFV:
Также доступен расширенный тест, позволяющий проводить дифференциальную диагностику наиболее часто встречающихся аутовоспалительных заболеваний:
Так как ССЛ является аутосомно-рецессивным заболеванием, то для подтверждения диагноза требуется обнаружение гомозиготной мутации либо двух гетерозиготных мутаций. Наличие одной гетерозиготной мутации повышает вероятность диагноза "ССЛ" и оправдывает начало терапии препаратами колхицина. Отсутствие мутаций во 2, 3, 5, 10 экзонах гена MEFV на 98% исключает диагноз ССЛ.
У пациентов с семейной средиземноморской лихорадкой, нейтрофильными дерматозами и холодовыми периодическими синдромами отмечается выраженное повышение уровня кальпротектина в крови, особенно во время приступов, что делает возможным использование его в качестве маркера активности заболевания:
Как подтвердить диагноз семейной средиземноморской лихорадки?
⌄Для подтверждения диагноза семейной средиземноморской лихорадки используется молекулярно-генетическое исследование (экзоны 2,3,5,10 гена MEFV методом секвенирования или полногенное исследование гена MEFV методом NGS). Диагноз критериальный и учитывает как клинические данные, так и данные молекулярно-генетического исследования.
Чем опасна семейная средиземноморская лихорадка без лечения?
⌄Главное осложнение — AA-амилоидоз с поражением почек, развивающийся у 60-75% пациентов без лечения и приводящий к хронической почечной недостаточности.
Семейная средиземноморская лихорадка встречается в славянской популяции?
⌄Да, семейная средиземноморская лихорадка (ССЛ) встречается как в славянской популяции, так и у коренных народов Северной Европы, однако в этих группах она считается крайне редким (орфанным) заболеванием. Исторически ССЛ считалась болезнью строго определенных этносов (армян, турок, арабов, евреев), происходящих из Средиземноморья. Современная генетика и миграционные процессы показали, что мутации гена MEFV присутствуют по всему миру. Однако если распространённость ССЛ среди армян, евреев-сефардов, турок, арабов составляет от 1:500 до 1:1000, то в славянской популяции она намного ниже. Например, масштабные исследования в Центральной Европе (в частности, в Словакии) выявили реальную распространенность на уровне примерно 1 случай на 48 000 населения.
1) Семейная средиземноморская лихорадка (Наследственный семейный амилоидоз). Клинические рекомендации МЗ РФ. 2023 г.
2) Ватутин Н.Т., Смирнова А.С., Эль-Хатиб М.А. Семейная средиземноморская лихорадка: обзор рекомендаций EULAR, 2016. Архивъ внутренней медицины. 2016;6(6):5-11. https://doi.org/10.20514/2226-6704-2016-6-6-5-11
3) Federici S, Sormani MP, Ozen S et al. Paediatric Rheumatology International Trials Organisation (PRINTO) and Eurofever Project. Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis. 2015 May;74(5):799-805. doi: 10.1136/annrheumdis-2014-206580
4) Gattorno M, Hofer M, Federici S et al. Eurofever Registry and the Paediatric Rheumatology International Trials Organisation (PRINTO). Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. 2019 Aug;78(8):1025-1032. doi: 10.1136/annrheumdis-2019-215048
5) Demir, S., Günel, İ.E., Özen, S. (2019). Familial Mediterranean Fever. In: Efthimiou, P. (eds) Auto-Inflammatory Syndromes. Springer, Cham. https://link.springer.com/chapter/10.1007/978-3-319-96929-9_9
6) Cekin N, Akyurek ME, Pinarbasi E, Ozen F. MEFV mutations and their relation to major clinical symptoms of Familial Mediterranean Fever. Gene. 2017 Aug 30;626:9-13. doi: 10.1016/j.gene.2017.05.013
7) Wang HH. MEFV gene mutation spectrum in patients with familial mediterranean fever. Pediatr Neonatol. 2023 Mar;64(2):107-108. doi: 10.1016/j.pedneo.2023.02.001